




Autoantibodies in Scleroderma

Scleroderma, also known as systemic sclerosis (abbreviated hereafter as SSc), remains to this day a disease of unknown cause. However, research has made considerable progress in understanding the mechanisms involved in the lesions caused by the disease, both in the skin and in the internal organs. The study of the mechanisms of a disease is called “pathophysiology”. The mechanisms that contribute to pathophysiology are referred to as “pathogenic”.
FOUR MAJOR MECHANISMS OF SCLERODERMA
Four major pathogenic mechanisms are recognized in the pathophysiology of SSc.
1. First, there is a dysfunction of the immune system that causes it to attack the individual’s own body. This explains why SSc is considered an “autoimmune” disease.
2. There is also microvascular damage, meaning that it targets the small blood vessels. Raynaud’s phenomenon (fingers changing colour when exposed to cold temperatures) present in almost all SSc patients, and which is so often both the first symptom and the first sign of the disease, is the typical manifestation of this vascular damage.
3. There is also inflammation. Although it is often underestimated in SSc, research has shown its crucial importance, to the point that “without inflammation, there is no fibrosis”.
4. Finally, fibrosis (or sclerosis, which gives its name to SSc) is the ultimate result of the previous intertwined mechanisms, both in the skin and in the internal organs.
As we can see, the pathophysiology of SSc is complicated. These pathogenic mechanisms have been the subject of intensive research in order to understand their interrelationships and to find “weak points” in the pathophysiological cascades that may become new therapeutic targets. In this article, we will focus on the dysregulation of the immune system and, in particular, on the specific antibodies that are typically found in the blood of SSc patients. Other important and complex aspects of immune system dysfunction in SSc (such as cellular immunity, innate immunity and signaling) will not be discussed here.
IMMUNE SYSTEM DYSFUNCTION IN SCLERODERMA
One of the primary functions of the immune system is to protect the individual from infection. Normally, the immune system does this job extraordinarily well, even though every day each individual is exposed to countless potentially dangerous microbes both in the human body itself and in the environment.
However, there are times when the immune system goes haywire and produces antibodies that attack the individual instead of protecting them. This is what happens in several autoimmune diseases, and the antibodies produced are called “autoantibodies”.
Indeed, SSc is characterized by highly specific autoantibodies in the blood of affected individuals. By “specific” we mean that these autoantibodies are not seen in other diseases.
AUTOANTIBODIES CONTRIBUTE TO THE DIAGNOSIS OF SCLERODERMA
There are four main autoantibodies, referred to as “classic”,
which are named as follows:
- anti-centromere
- anti-topoisomerase I
- anti-RNA polymerase III
- anti-Th/To
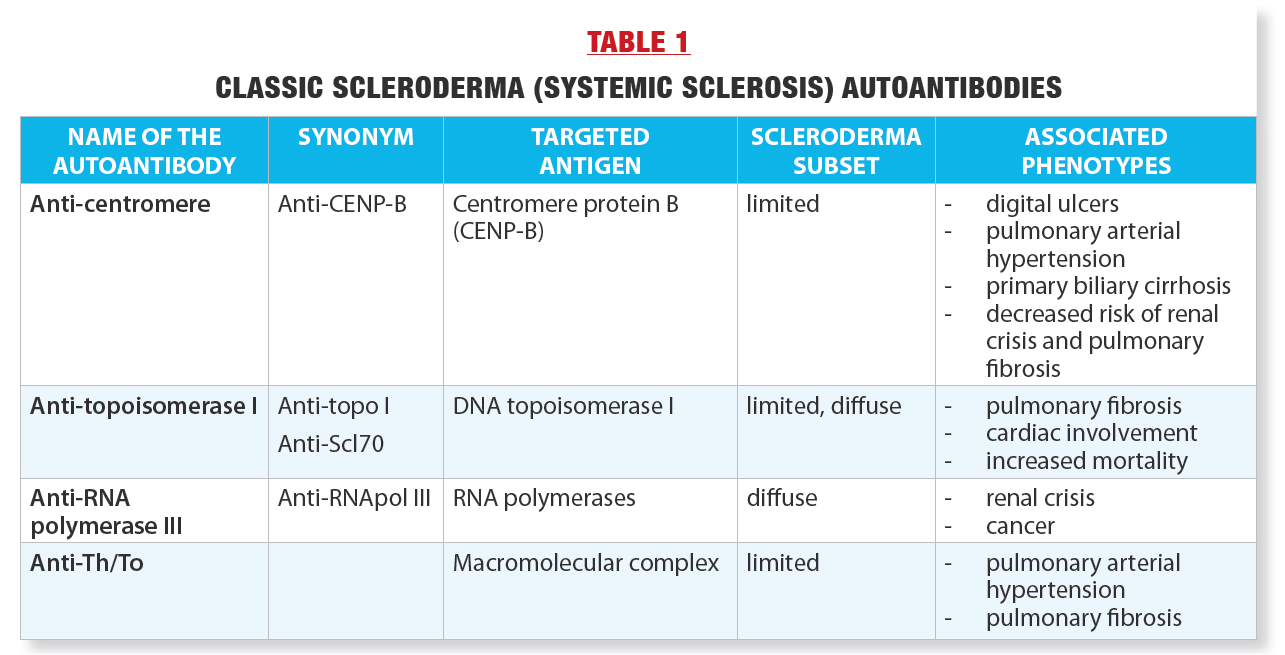
These autoantibodies, their synonyms, and the molecules against which they are directed (antigens) are listed in Table 1. In general, the SSc autoantibodies are mutually exclusive: in a given individual, only one of the four autoantibodies will be present. Several other less common autoantibodies have been described in recent years that are also associated with SSc, but because of their rarity and the fact that their significance still needs to be better defined, they will not be discussed here.
In fact, the four classic autoantibodies are so specific to SSc that there are now laboratory tests for their detection. In an individual with certain signs of SSc, such as Raynaud’s phenomenon and skin thickening, the presence of a high titer (a significant amount) of any of these autoantibodies in the blood helps to support the diagnosis of the disease.
Thus, after a detailed questionnaire and physical examination, the rheumatologist or other medical specialist will request a blood test for these autoantibodies if they suspect a diagnosis of SSc. The diagnostic workup is usually completed by a capillaroscopy (microscopic examination of the small blood vessels around the nails) and also by tests to evaluate the internal organs, such as a CT scan of the lungs and lung function tests to look for pulmonary fibrosis.
It should be noted that the mere presence of one of the four autoantibodies in the blood does not definitively establish a diagnosis of SSc: there must be other clinical manifestations to support this diagnosis.
EACH AUTOANTIBODY IS ASSOCIATED WITH SPECIFIC CLINICAL MANIFESTATIONS
A striking feature of the four autoantibodies is that each is associated with particular clinical manifestations, called “phenotypes,” as shown in Table 1.
For instance, anti-centromere autoantibodies are associated with the limited cutaneous form of SSc, in which skin thickening is typically limited to the fingers and forearms and life expectancy is longer. Patients with these autoantibodies appear to be at lower risk of some potentially serious manifestations of SSc, such as renal crisis (sudden cessation of kidney function accompanied by extremely high blood pressure that can lead to death) or lung fibrosis (pulmonary fibrosis).
On the other hand, anti-centromere autoantibodies are associated with the eventual occurrence (often after a long evolution) of increased pressure in the arteries of the lungs (pulmonary hypertension) which can be become life-threatening over time.
Anti-centromere autoantibodies are common in the Quebec scleroderma population of French-Canadian origin. In fact, in our study of SSc in French Canada involving 309 affected individuals, more than 40% of the patients were anticentromere carriers (1).
In comparison, anti-topoisomerase I autoantibodies are less common, occurring in about 15% of our population with SSc. However, their detection is important because they are associated with higher risk of potentially severe pulmonary fibrosis and also heart (myocardial) damage.
In a study comparing the life expectancy of our patients separated according to the four autoantibodies, the survival ten years after diagnosis was worse in patients with anti-topoisomerase I (67% survival rate) or anti-RNA polymerase III (85%) autoantibodies, and better in those with anti-centromere (90%) or anti-Th/To (100% autoantibodies).
Thus, testing for SSc autoantibodies is not only useful for diagnosing SSc but also provides important clinical information for predicting the associated phenotype and potential disease course. The physician can then individually tailor the follow-up and intensity of treatments according to the risks identified by the autoantibody present.
PRESENCE OF AUTOANTIBODIES FROM THE ONSET OF SCLERODERMA – PRESCLERODERMA
Another characteristic of the four autoantibodies is that they are present in the blood early in SSc, at the onset of the first signs of the disease. This concept was derived from a small number of observations made in the 20th century involving individuals with Raynaud’s phenomenon only and carrying anti-centromere or anti-topoisomerase I autoantibodies who subsequently developed SSc. However, this concept had never been validated in a large number of subjects with long-term follow-up. Our research team, therefore, undertook a 20-year prospective study of 586 adult individuals with isolated Raynaud’s phenomenon (without any other manifestation of SSc or other autoimmune diseases) to see who would develop SSc over time(2). These individuals all had capillaroscopy at baseline and also had blood drawn for the four SSc autoantibodies.
Of course, given that isolated Raynaud’s phenomenon is common in the adult female population in Quebec, most of these individuals did not develop SSc. However, 74 individuals, or 12.6% of the 586 participants, developed SSc at follow-up.
At follow-up, 80% of individuals who had one of the four SSc autoantibodies at baseline and an abnormal capillaroscopy developed SSc(2). These individuals were 60 times more likely to develop SSc than those who tested negative. The time interval for developing SSc ranged from a few months to a few years.
This study, therefore, identified factors that predict a high risk of progression to SSc in individuals with isolated Raynaud’s phenomenon(2). In addition, the study demonstrated the existence of an early phase of SSc, now referred to as prescleroderma,during which the disease appears to be incubating but cannot be formally diagnosed by physicians.
This novel concept opens the door to research projects aimed at better understanding the pathophysiology of this incubation period and the mechanisms leading to the progression to definitive SSc, with the hope of identifying new preventive therapeutic targets.
DO AUTOANTIBODIES CONTRIBUTE TO SCLERODERMA LESIONS?
As we have seen, the presence of highly specific autoantibodies is compelling evidence of immune system involvement in SSc. Moreover, each autoantibody is associated with a specific SSc phenotype (Table 1). Finally, SSc autoantibodies are present as far back as possible, presumably from the onset of Raynaud’s phenomenon(2).
These data inevitably raise the question: is there evidence that these autoantibodies themselves contribute to the pathophysiology of SSc lesions? In other words, are SSc autoantibodies pathogenic?
In 2019, our research team was approached by the editors of the Journal of Scleroderma and Related Disorders, the only medical journal dedicated to scientific research in SSc. Drs. M. Matucci-Cerinic (University of Florence) and M. Kuwana (Nippon Medical School, Tokyo) asked us to prepare an article aimed at answering this very thorny question. The question is not theoretical: if evidence were to show a pathogenic role of SSc autoantibodies, would it not be of great interest to develop new treatments targeting these autoantibodies to block their deleterious effects?
Our team at the Centre hospitalier de l’Université de Montréal, composed of Sabrina Hoa, MD, Roger Yang, MD, Martial Koenig, MD, and the undersigned, set out to review all the data accumulated over the past 40 years. The result was a 27-page article, with 182 references, published in 2020(3). Here are the two main conclusions.
The first conclusion is that in order to assert the pathogenic role of an autoantibody in SSc (or in any systemic autoimmune disease), one must first establish rigorous scientific criteria for pathogenicity. We have therefore proposed 7 rigorous criteria that should ideally be present to state without any doubt that an autoantibody is pathogenic. These criteria are presented in Table 2.

AS WE CAN SEE IN TABLE 2, THE SCIENTIFIC BAR HAS BEEN SET HIGH!
In a second phase, the team screened all published articles on the pathogenic role of autoantibodies in SSc and catalogued them for each of the 7 criteria according to the following grading scale:

Finally, using these assessments, a verdict was reached as to whether the pathogenic role of autoantibodies was definitive, probable, possible, or whether the data were insufficient. Table 3 shows the results. Of the four classic SSc autoantibodies, only anticentromere and anti-topoisomerase I autoantibodies have been extensively studied for their pathogenic role. The second conclusion was that there is indeed some evidence of pathogenicity for these two autoantibodies and therefore their pathogenic role is possible(3). As shown in Table 3, the evidence for a pathogenic role is strongest for anti-topoisomerase I. Thus, further research is needed to better understand how these autoantibodies contribute to SSc lesions.

CONCLUSION
As we have seen, the four classic SSc autoantibodies are essential for the diagnosis of the disease and are useful in predicting its manifestations, course and associated life expectancy. The high specificity of these autoantibodies for SSc, their association with a particular phenotype and their presence from the onset of the disease suggest that they play a pathogenic role, the demonstration of which according to rigorous scientific criteria is well underway and needs to be completed.
Moreover, the remarkable association of these autoantibodies with SSc suggests that they are directly and intimately linked to the cause of the disease, the identity of which remains a mystery to this day.

ACKNOWLEDGMENTS
The author would like to thank Martial Koenig, MD and
Sabrina Hoa, MD for reviewing the text.
Special thanks to Scleroderma Quebec for its unwavering
support of the Research Chair since 2003.
REFERENCES :
1. Scussel-Lonzetti L, Joyal F, Raynauld JP, Roussin A, Rich É, Goulet JR, Raymond JR, Senécal JL. Predicting mortality in systemic sclerosis : analysis of a cohort of 309 French Canadian patients with emphasis on features at diagnosis as predictive factors for survival. Medicine (2002) 81: 154-167. doi: 10.1097/00005792-200203000-00005.
2. Koenig M, Joyal F, Fritzler MJ, Roussin A, Abrahamowicz M, Boire G, Goulet JR, Rich É, Grodzicky T, Raymond Y, Senécal JL. Autoantibodies and microvascular damage are independent predictive factors for the progression of Raynaud’s phenomenon to systemic sclerosis. A 20-year prospective study of 586 patients with validation of proposed criteria for early systemic sclerosis. Arthritis Rheum (2008) 58: 3902-3912. doi:10.1002/art.24038.
3. Senécal JL, Hoa S, Yang R, Koenig M. Pathogenic roles of autoantibodies in systemic sclerosis: current understandings in pathogenesis. J Scleroderma Relat Disord (2020) 5:103-129. doi.org/10.1177/2397198319870667.
March 2023
